
Codice esenzione RCG040
La cistinuria è una malattia congenita congenita caratterizzata da un metabolismo della cistina difettoso che provoca la formazione di calcoli di cistina. Tra il gruppo eterogeneo di malattie dei calcoli renali, la cistinuria è l’unico disturbo causato esclusivamente da mutazioni genetiche.
Finora sono stati identificati due geni responsabili della cistinuria: SLC3A1 (cromosoma 2p21) codifica la subunità pesante rBAT di un trasportatore renale b 0, + mentre SLC7A9 (cromosoma 19q12) codifica la sua sub-unità leggera interagente b 0, + AT. Le mutazioni di SLC3A1 sono generalmente associate a una modalità ereditaria autosomica recessiva mentre SLC7A9le varianti comportano un’ampia variabilità clinica anche all’interno della stessa famiglia. Il tasso di rilevazione delle mutazioni in questi geni è superiore all’85%, ma è influenzato dall’origine etnica di un paziente e dal significato patofisiologico delle mutazioni. Oltre alla cistinuria isolata, sono stati segnalati pazienti che soffrono della sindrome ipotonia-cistinuria portando delezioni tra cui almeno SLC3A1 e PREPL geni in 2p21. Mediante ampi studi di screening molecolare in un’ampia coorte di pazienti è stato identificato un ampio spettro di mutazioni, molte di queste varianti sono state analizzate funzionalmente e quindi hanno permesso di approfondire la patologia della malattia e il traffico renale di cistina e aminoacidi dibasici . Nella nostra recensione riassumeremo le attuali conoscenze sulle basi fisiologiche e genetiche della cistinuria come causa innata di calcoli renali e l’applicazione di queste conoscenze nelle strategie di test genetici.
Revisione
L’incidenza delle malattie dei calcoli renali è aumentata negli ultimi decenni nei paesi industriali a quasi l’1,5% nel 2000, circa il 5% di tutte le donne e il 12% degli uomini svilupperanno un calcolo renale una volta nella vita .L’incidenza nell’infanzia è inferiore ed è stata stimata in circa lo 0,15% (per la revisione: [ 2 ]). È interessante notare che circa il 40% dei bambini con calcoli renali ha una storia familiare positiva mentre l’urolitiasi negli adulti spesso si verifica sporadicamente.
In caso di formazione di calcoli, l’analisi qualitativa è una delle misure diagnostiche più importanti: calcio e ossalato sono i principali componenti lapidei nella popolazione europea rilevabili in oltre il 75% dei pazienti, mentre fosfato, cistina, purina e altri calcoli sono spesso raro (per revisione: [ 2 ]). Molti fattori litogenici e inibitori sono coinvolti nell’eziologia della formazione di calcoli che è significativamente influenzata dall’assunzione di liquidi e da fattori dietetici.
In effetti, una predisposizione genetica può essere osservata in molti disturbi associati ai calcoli renali, ma la cistinuria è l’unica entità causata esclusivamente da mutazioni genetiche. Tra gli adulti, i calcoli di cistina rappresentano solo l’1-2% di tutti i pazienti con nefrolitiasi urinaria, ma nei bambini il 6-8% dei pazienti soffre di calcoli di cistina [ 3 ].
La cistinuria è caratterizzata dal riassorbimento difettoso di cistina, lisina, ornitina e arginina nella membrana del bordo del pennello del tubulo renale prossimale (segmento S3) e nelle cellule epiteliali del tratto gastrointestinale (per revisione: [ 4 ]) . Sebbene tutti e quattro gli amminoacidi raggiungano elevate concentrazioni urinarie, solo la risultante ipersecrezione urinaria della cistina porta alla precipitazione nel tubulo distale e alla formazione di calcoli di cistina a causa della sua bassa solubilità a basso pH. Nei pazienti con grandi delezioni genomiche che colpiscono il gene SLC3A1 e almeno il gene PREPL neighboured in 2p21, l’urolitiasi è inoltre associata a ipotonia e ulteriori sintomi clinici (vedere sotto).
Risultati clinici e classificazione
La prima diagnosi di cistinuria si basa comunemente sulla scoperta di calcoli di cistina che mostrano tipicamente cristalli di cistina caratteristici. I cristalli sono generalmente esagonali, traslucidi e bianchi. Dopo la rimozione, le pietre possono essere di colore rosa o giallo, ma successivamente diventano verdastre a causa dell’esposizione all’aria. I cristalli di cistina sono visibili nel 17-25% dei campioni di urina di pazienti con cistinuria (per revisione: [ 2 ]). Le pietre possono essere identificate da un test nitroprussiato al cianuro positivo. La maggior parte delle pietre sono eccezionalmente formate da cistina, ma si può osservare una composizione mista. La diagnosi può essere confermata dalla determinazione dell’escrezione urinaria di aminoacidi: in caso di cistinuria l’escrezione urinaria di cistina è in genere aumentata a> 1000 μmoL / g di creatinina [ 5].
Tuttavia, a causa dell’espressione incompleta dei trasportatori di aminoacidi renali, è possibile osservare una cosiddetta cistinuria neonatale transitoria negli eterozigoti della cistinuria, pertanto la diagnosi prima dei 4 anni di vita deve essere effettuata con cautela. [ 6 ]
A causa della loro composizione, la frammentazione delle pietre di cistina da parte della litotrissia extracoporale è spesso difficile e le pietre più grandi richiedono generalmente il posizionamento della nefrostomia percutanea e la rimozione. I sintomi episodici delle pietre rendono necessarie ripetute rimozione delle pietre. In effetti, il disturbo provoca gravi danni ai reni e agli organi circostanti e in alcuni rari casi la morte se non trattata correttamente. L’attuale trattamento della cistinolitiasi è attualmente focalizzato sulla prevenzione della formazione di calcoli riducendo l’escrezione e la concentrazione di cistina e riducendo la cistina alla cisteina più solubile (per la revisione: [ 4 ]). Tuttavia, le terapie causali non sono ancora state sviluppate nonostante la crescente conoscenza dell’eziologia fisiologica della malattia.
A fini clinici, la classificazione della cistinuria si basa sul fenotipo urinario degli eterozigoti obbligati (cioè i genitori di pazienti con un corso classico di cistinuria) e sono stati distinti tre tipi di cistinuria [ 7 , 8 ]. Mentre gli eterozigoti di tipo I espellono la cistina a livelli normali, gli eterozigoti di tipo II e III mostrano un’escrezione altamente o moderatamente elevata. Tuttavia, dopo l’identificazione delle mutazioni genetiche che predispongono alla cistinuria, non è stato possibile stabilire una correlazione tra l’entità dell’iperaminoaciduria e la mutazione negli eterozigoti di tipo II / III. Pertanto, entrambi i tipi II e III sono stati riassunti come non di tipo I [ 9]. Gli eterozigoti non di tipo I mostrano una ipersecrezione urinaria variabile di cistina e aminoacidi dibasici, in alcuni portatori è stata segnalata la formazione di calcoli [ 10 ]. Di conseguenza, la cistinuria non di tipo I può essere considerata una malattia autosomica dominante con penetranza incompleta per la litiasi da cistina, mentre la cistinuria di tipo I segue principalmente un tratto autosomico-recessivo. Inoltre, sono stati riportati pazienti con cistinuria mista, con alleli sia di tipo I che non di tipo I (per revisione: [ 4 ]).
Nella maggior parte dei pazienti con cistinuria, la formazione di calcoli si verifica entro i primi due decenni di vita [ 11 ], ma è stata segnalata un’ampia variazione intrafamiliale della malattia. I maschi sono colpiti più frequentemente e gravemente delle femmine e i maschi hanno un numero maggiore di calcoli. Oltre l’80% dei pazienti sviluppa le prime pietre entro i primi due decenni, ma le pietre possono formarsi a qualsiasi età. Nei pazienti maschi si può osservare una precedente formazione di calcoli rispetto alle femmine. Circa il 6% dei pazienti non forma calcoli [ 12 ].
Goodyer et al. [ 5 ] hanno osservato una preponderanza di una manifestazione precoce della formazione di calcoli nei pazienti omozigoti di tipo I, mentre i pazienti omozigoti di tipo I e quelli di tipo misto hanno sviluppato pietre più avanti nella vita. Tuttavia, questa correlazione non ha potuto essere confermata in pazienti molecolari [ 13 ].
Frequenza
La cistinuria è una malattia globale con prevalenze specifiche della popolazione, la sua prevalenza complessiva è stata stimata in 1: 7000 nei neonati [ 14 ]. Varia tra le diverse popolazioni: la più alta frequenza è stata osservata tra gli ebrei libici con un tasso di 1: 2.500, negli americani il tasso è di 1: 15.000 e in Svezia 1: 100.000 [ 14 , 15 ]. In popolazioni specifiche può quindi essere considerato come uno dei più comuni disturbi autosomici recessivi paragonabile solo alla fibrosi cistica. Tuttavia, a causa di queste informazioni limitate sulla prevalenza della cistinuria nella popolazione generale e sull’ampio spettro di mutazioni, la frequenza dei portatori di cistinuria può essere solo stimata. Il tasso di portatore più alto di 1:25 negli ebrei libici è stato identificato da Sidi et al. [16 ]. Nella popolazione svedese, p.Met467Thr si verifica nello 0,5% della popolazione generale [ 17 ].
I geni SLC3A1 e SLC7A9 e il sistema di trasporto degli aminoacidi b 0, +
Il coinvolgimento del gene SLC3A1 (OMIM 104614) nell’eziologia della cistinuria potrebbe essere stabilito nel 1994 stabilendo il collegamento della malattia a 2p e identificando le prime mutazioni nei pazienti con cistinuria [ 18 , 19 ]. Ulteriori studi di collegamento genetico hanno indicato che non tutte le famiglie di cistinuria erano causate da difetti di SLC3A1 e che il secondo gene della cistinuria poteva essere localizzato nel cromosoma 19q13 [ 20 , 21 ]. Nel 1999 sono state riportate le prime mutazioni del gene SLC7A9 [ 9 ].
Il gene SLC3A1 si estende per ~ 46 kb e include 10 esoni codificanti [ 22 , 23 ]. La trascrizione più lunga è lunga 2989 bp, la proteina rBAT risultante è composta da 685 aminoacidi. Il gene SLC7A9 comprende 12 esoni, 11 dei quali codificanti. La trascrizione ha 1772 bp, la proteina b 0, + AT è composta da 487 aminoacidi.
I due geni codificano le due sub-unità del trasportatore eterometrico renale di aminoacidi b 0, + : la sub-unità pesante rBAT ( SLC3A1 ) e la sub-unità leggera b 0, + AT ( SLC7A9 )) (Figura 1 ) (per revisione: [ 4 ] ), entrambi collegati da un ponte disolfuro conservato. La sub-unità pesante media la localizzazione dell’ holotransporter sulla membrana plasmatica mentre la sub-unità leggera comprende il componente catalitico del trasportatore.

Localizzazione cellulare e funzione di rBAT ( SLC3A1 ) eb 0, + AT ( SLC7A9 ).
Immagine a dimensione intera
Il trasportatore di aminoacidi rBAT / b 0, + è espresso nella membrana apicale delle cellule epiteliali dei segmenti S1-S2 del tubulo prossimale e dell’intestino tenue. Tramite il trasportatore etero dimero gli amminoacidi dibasici e la cistina vengono scambiati con altri amminoacidi neutri in una stechiometria 1: 1. rBAT / b 0, + AT rappresenta il principale meccanismo di trasporto tubulare della cistina e rappresenta> 90% dell’assorbimento renale della cistina.
Come risultato delle sue funzioni biologiche, le mutazioni di SLC3A1 / rBAT hanno forti effetti di traffico, sia per mancanza di assemblaggio con b 0, + AT o per mancata oligomerizzazione con conseguente degradazione (per revisione: [ 4 ]). b 0, + Le mutazioni AT causano una perdita di funzionalità del sistema trasportatore a causa di piegatura, traffico, etero dimerizzazione, attività di trasporto o riconoscimento del substrato difettosi (per revisione: [ 4 ]).
Genetica e spettro delle mutazioni
Come accennato in precedenza, la classificazione della cistinuria era storicamente basata sul modello di escrezione urinaria di eterozigoti. Dopo l’identificazione delle basi molecolari della malattia, è stata suggerita una nuova classificazione: la cistinuria di tipo I ereditata in modo autosomico che è principalmente causata dalle mutazioni SLC3A1 e la cistinuria non di tipo I dominante autosomica incompleta associata alle varianti di SLC7A9 . Tuttavia, l’identificazione di entrambe le mutazioni SLC3A1 che causano cistinuria non di tipo I e le mutazioni recessive SLC7A9 portano a una nuova classificazione strettamente molecolare [ 11 ] che non include il fenotipo biochimico: la cistinuria di tipo A rappresenta SLC3A1mutazioni e cistinuria di tipo B include mutazioni SLC7A9 . Pertanto, possono essere delineati tre genotipi: AA, BB e la cistinuria mista AB.
In studi di screening di grandi dimensioni, sono state riportate oltre 130 varianti patogene in SLC3A1 e quasi 100 mutazioni in SLC7A9 (per revisione: [ 4 , 24 ]). Le varianti osservate coprono l’intero spettro delle mutazioni, che vanno dalle mutazioni senza senso, missense, splicing e frame-shift agli squilibri interi e multi-esone. Il ruolo significativo dei riarrangiamenti genomici di grandi dimensioni è diventato evidente con l’identificazione delle prime eliminazioni / duplicazioni in SLC3A1 [ 25 ] e la caratterizzazione della duplicazione relativamente frequente che colpisce gli esoni da 5 a 9 (dupE5E9; c.891 + 1524_1618-1600dup) in SLC3A1 [ 4 , 24] che rappresentano almeno l’11% degli alleli SLC3A1 mutati (Tabella 1 ). Bisceglia et al. Hanno recentemente riportato uno schermo sistematico per gli squilibri esoni / multi-esoni. [ 24 ] applicando MLPA (amplificazione dipendente dalla sonda per legatura multipla): potrebbero dimostrare che i riarrangiamenti di grandi dimensioni contribuiscono in modo significativo allo spettro delle mutazioni in entrambi i geni della cistinuria in Italia.
Tabella 1 Panoramica sulle mutazioni di cistinuria più frequenti in a) SLC3A1 eb ) SLC7A9 in diversi gruppi etnici
Tavolo a grandezza naturale
Nei singoli pazienti, possono essere rilevate tre mutazioni patogene [ 27 , 31 ]. In effetti, l’identificazione di tre varianti recessive nello stesso gene è una scoperta rara ma è un’osservazione ben nota anche da altri disturbi autosomici recessivi, ad esempio fibrosi cistica o malattia renale policistica autosomica recessiva. Ciò significa che almeno due mutazioni sono localizzate sullo stesso allele e ciò potrebbe portare a risultati falsi positivi o falsi negativi nella diagnostica del corriere.
SLC3A1
Come accennato in precedenza, le mutazioni in SLC3A1 sono generalmente associate alla cistinuria di tipo I, ma esistono le eccezioni dupE5E9 [ 10 ]; portatori di eterozigoti di queste mutazioni mostrano un aumento del modello di escrezione di cistina urinaria.
Le mutazioni sono distribuite su tutto il gene, interessando tutti gli esoni e tutti i domini funzionali (Figura 2 ). Tra le mutazioni SLC3A1 attualmente note p.Met467Thr è generalmente la più frequente, rappresentando circa il 30% degli alleli SLC3A1 noti e rilevabile in quasi tutti i gruppi etnici (Tabella 1 ).
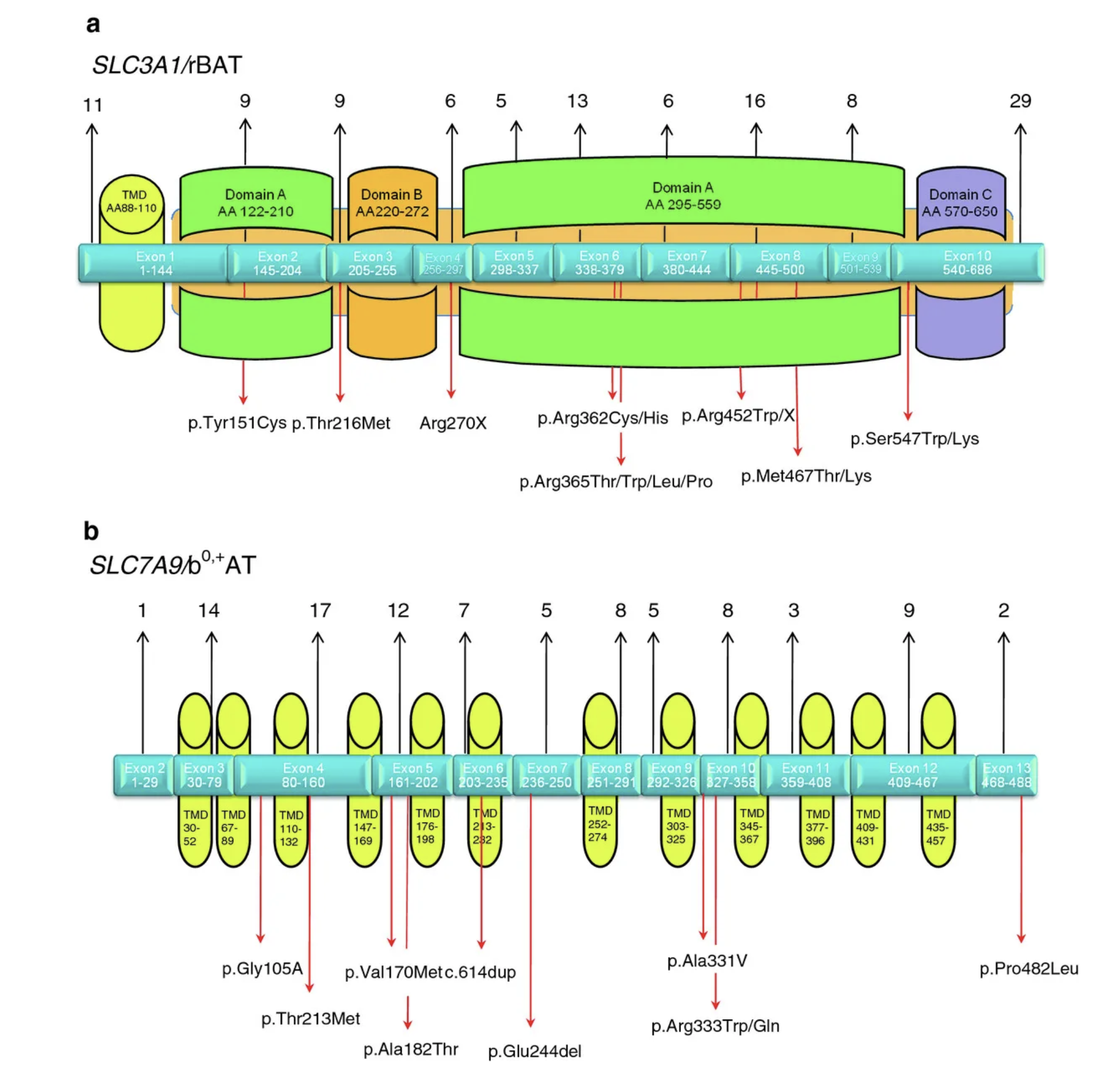
Struttura genomica e funzione putativa delle regioni proteiche codificate dei geni della cistinuria: il numero totale di mutazioni descritte finora per ciascun esone è mostrato sopra, la localizzazione delle mutazioni più frequenti è mostrata sotto la struttura dell’esone. a) SLC3A1 / rBAT (dominio transmembranico TMD; la maggior parte della proteina è costituita da un ectodominio (marrone chiaro) di tre domini AC, per ulteriori dettagli si veda [ 4 ]). b) SLC7A9 / b 0, + AT (dominio transmembrana TMD).
Immagine a dimensione intera
La sostituzione amminoacidica p .Thr216Met nell’esone 3 è la seconda variante frequente (~ 13% di tutti gli alleli SLC3A1 identificati ), ma mostra una preponderanza nei pazienti dell’Europa sud-orientale (per revisione: [ 4 , 25 ]) e quindi riflette l’effetto dell’origine etnica sulla distribuzione delle mutazioni della cistinuria.
Per alcune varianti si può osservare un’influenza etnica: è per lo più evidente per la mutazione p.Arg270X che è stata identificata nel 73% dei pazienti ebrei Ashkenazi e rappresenta l’11% delle mutazioni SLC3A1 conosciute in Nord America (Tabella 1 ). La mutazione p.Tyr151Cys è stata osservata principalmente nel Nord Europa [ 34 ]. La duplicazione dupE5E9 potrebbe essere avvenuta da un fondatore tedesco, tuttavia i dati sono limitati in quanto in passato i test miranti a squilibri genomici più grandi non venivano applicati abitualmente per la diagnostica della cistinuria.
SLC7A9
Le conseguenze funzionali delle mutazioni di SLC7A9 sono generalmente più ampie di quelle in SLC3A1 . Per molte mutazioni di SLC7A9 , si può osservare un’influenza autosomica dominante rispetto ai modelli di aminoacidi urinari, ma la penetranza è incompleta rispetto alla fomazione di calcoli. Di conseguenza, una correlazione genotipo-fenotipo è semplicemente possibile e si può osservare un ampio spettro biochimico e clinico intrafamiliale per la stessa mutazione.
Simile a SLC3A1 , le mutazioni in SLC7A9 possono essere rilevate in tutti gli esoni del gene (Figura 1 ) ma ci sono punti caldi per le mutazioni negli esoni 3, 4, 5 e 6 (Tabella 1 ). Tra le mutazioni SLC7A9 , la sostituzione amminoacidica p.Gly105Arg rappresenta circa il 20% degli alleli identificati ed è presente in quasi tutti i gruppi etnici.
La seconda mutazione frequente p.Arg333Trp è rilevabile anche in tutto il mondo, mentre altre varianti sono spesso rare. È interessante notare che tre mutazioni mostrano una forte associazione etnica: p.Val170Met è limitato agli ebrei libici dove rappresenta tutti i pazienti; p.Pro482Leu colpisce> 84% degli alleli SLC7A9 identificati nella coorte giapponese di cistinuria [ 42 ]. La duplicazione c.614dupA è preponderante nei pazienti spagnoli, rappresentando il 29% degli alleli SLC7A9 e probabilmente proveniente da un fondatore delle Asturie (per la revisione: [ 4 ]).
Nel gruppo della rara mutazione SLC7A9 , la variante p.Thr123Met deve essere menzionata separatamente in quanto illustra l’ampio fenotipo fisiologico e clinico delle mutazioni SLC7A9 . I modelli di escrezione di amminoacidi possono variare da cistinuria quasi normale a isolata a ipersecrezzione di cistina e amminoacidi dibasici nei portatori di p.Thr123Met [ 10 , 43 ].
Sindrome ipotonia-cistinuria e microdelezioni 2p21
Le più grandi delezioni che colpiscono SLC3A1 potrebbero causare la cosiddetta sindrome ipotonia-cistinuria (HCS; OMIM 606407). Questo disturbo congenito autosomico recessivo è associato a eliminazioni di almeno i geni SLC3A1 e PREPL sul cromosoma 2p21. Le principali caratteristiche cliniche includono ipotonia generalizzata alla nascita, insufficienza a prosperare, ritardo della crescita e cistinuria. Nel frattempo sono stati segnalati 13 pazienti con HCS, tutti erano omozigoti per le delezioni in 2p21 [ 44 , 45 ]. Finora sono state identificate cinque diverse eliminazioni di HCS, due delle quali (eliminazioni “A” e “B”) sono state trovate per essere distribuite a livello globale. Nonostante le loro diverse dimensioni che vanno da ~ 38 a ~ 127 kb, tutti influenzano (almeno in parte) il Geni SLC3A1 e PREPL che provocano delezioni omozigoti funzionalmente di entrambi i geni. Un ulteriore paziente con cistinuria come unica caratteristica clinica è stato rilevato dallo screening dei pazienti con cistinuria per le mutazioni di SLC3A1 ; il paziente era eterozigote composto per una delezione di SLC3A1 / PREPL e una delezione in SLC3A1 che interessava gli esoni da 1 a 7 [ 40 ]. Contrariamente all’associazione delle mutazioni di SLC3A1 con la cistinuria, non sono state ancora riportate varianti limitate a PREPL che causano un fenotipo di “ipotonia isolata”. Tuttavia, è stato ipotizzato che il fenotipo nei pazienti con HCS possa essere attribuito alla mancanza di PREPL, una putativa oligopeptidasi serina con una funzione fisiologica attualmente sconosciuta [ 44 ].
Una seconda sindrome da micro delezione che colpisce anche 2p21 ma di dimensioni maggiori (~ 179 kb) viene definita sindrome da delezione 2p21 [ 46 ]. Questa cancellazione è stata rilevata in un grande pedigree beduino. I portatori di delezione omozigote in questa famiglia hanno mostrato HCS e caratteristiche aggiuntive tra cui convulsioni neonatali e un grave ritardo globale, indicando che la perdita di un terzo gene in 2p21, PPM1B , contribuisce anche allo spettro clinico in questa famiglia. Nel frattempo, sono stati descritti ulteriori pazienti con fenotipo intermedio tra HCS e sindrome da microdelezione 2p21 e delezioni di dimensioni intermedie [ 47 ].
Ulteriori geni candidati alla cistinuria
Considerando l’osservazione che in molti studi i tassi di rilevazione delle mutazioni in SLC3A1 o SLC7A9 non raggiungono il 100% e a causa della natura complessa del trasporto di aminoacidi renali, sono stati postulati il ruolo di ulteriori geni e fattori modificanti nell’eziologia della cistinuria. Tuttavia, le analisi di collegamento nelle famiglie di cistinuria non hanno indicato l’esistenza di più di due loci di cistinuria, 2p21 ( SLC3A1 ) e 19q13 ( SLC7A9 ), quindi la localizzazione di ulteriori geni che codificano le sub-unità trasportatrici di aminoacidi all’interno di questa regione era concepibile.
Un candidato nel 19q13 era SLC7A10 (ASC-1) che mostra un’elevata omologia con SLC7A9 ma diversi studi hanno escluso mutazioni patogene in questo gene [ 31 , 48 – 50 ]. Anche il sistema trasportatore di aminoacidi neutro ATB (0) ( SLC1A5 ) è localizzato nel 19q13, ma l’analisi delle mutazioni nelle famiglie di cistinuria con possibile collegamento con questa regione non ha fornito prove del contributo delle mutazioni SLC1A5 al decorso clinico [ 32 ].
La mancanza di mutazioni su singoli alleli può anche essere spiegata dal dominio fisiologico di alcuni alleli SLC7A9 , in questi casi non è necessaria una seconda mutazione per influenzare il fenotipo. Inoltre, è stato suggerito che a prima vista i polimorfismi a patogeni a singolo nucleotide (SNP) potrebbero predisporre un fenotipo di cistinuria in caso di co-occorrenza con altre mutazioni [ 28 , 51 ] e potrebbero fungere da modificatori.
Strategie di test genetici per la cistinuria
Lo sviluppo di test genetici per la cistinuria riflette la rapida evoluzione tecnologica negli ultimi due decenni. I test genetici molecolari sono stati inizialmente stabiliti dopo l’identificazione del cDNA rBAT e le prime mutazioni nel 1994. A quel tempo la struttura genomica di SLC3A1 non era noto e sono state stabilite singole PCR con successive digestioni di restrizione per mutazioni specifiche. Con la caratterizzazione della struttura genomica di entrambi i geni, sono stati applicati metodi di screening non specifici come SSCP (analisi del polimorfismo di conformazione a singolo filamento) per lo screening di grandi coorti di pazienti. Tuttavia, questi test erano limitati a causa della loro sensibilità e riproducibilità, pertanto il sequenziamento di Sanger è diventato il gold standard per l’analisi genetica molecolare in cistinuria. Per aggirare il problema che grandi riarrangiamenti non coperti dagli approcci di sequenziamento convenzionali potrebbero sfuggire al rilevamento, numerosi algoritmi quantitativi, come PCR quantitativa e amplificazione dipendente dalla sonda di legatura multipla (qPCR e MLPA), sono stati implementati in algoritmi diagnostici.
Sulla base della crescente conoscenza delle basi genetiche e biochimiche della cistinuria, prima del test genetico molecolare devono essere considerati i seguenti aspetti:
i dati biochimici e clinici potrebbero influenzare l’algoritmo diagnostico: nella maggior parte dei casi, i pazienti vengono sottoposti alla diagnosi clinica di urolitiasi. In questa situazione, l’attenta anamnesi familiare e i modelli di escrezione urinaria di aminoacidi da parte dei genitori potrebbero aiutare a decidere quale gene dovrebbe essere testato per primo: in caso di insorgenza sporadica e normale escrezione di aminoacido urinario parentale, SLC3A1 dovrebbe essere testato, in caso di positività storia familiare e iperaminoaciduria dei genitori, SLC7A9i test dovrebbero essere prioritari. Tuttavia la consanguineità potrebbe imitare un’eredità autosomica recessiva e le mutazioni in entrambi i geni mostrano un ampio intervallo osservabile di escrezione di cistina urinaria. Inoltre, i dati completi sono spesso difficili da ottenere o le informazioni dei membri della famiglia non sono disponibili.
Origine etnica : per popolazioni specifiche (ad es. Giappone, Svezia, Europa sud-orientale, Spagna) è possibile applicare un algoritmo mirato poiché questi gruppi mostrano mutazioni specifiche.
– procedura di test : mutazioni frequenti o specifiche della popolazione potrebbero essere testate mediante approcci di genotipizzazione mirati (ad es. saggi di restrizione). In caso di sequenziamento delle regioni di codifica e dei confini introne / esone, si potrebbe considerare di iniziare con gli esoni “hot spot” che ospitano mutazioni multiple e varianti frequenti (es. Esone 8 di SLC3A1 , esone 4 di SLC7A9 ). Nel caso in cui nessuna di queste mutazioni frequenti sia rilevabile, le sequenze di codifica totali di entrambi i geni devono essere sequenziate. Dopo l’esclusione delle mutazioni esoniche, dovrebbero seguire i test quantitativi poiché le duplicazioni / delezioni intrageniche contribuiscono in modo significativo allo spettro delle mutazioni nella cistinuria [ 24]. In caso di fenotipi insoliti, il cariotipo molecolare con microarrays del DNA è uno strumento appropriato per identificare grandi squilibri genomici come riportato per l’HCS [ 44 ]. Questa analisi per gradi può essere interrotta dopo l’identificazione di due varianti patogene che spiegano il decorso clinico del paziente, sebbene la presenza di una terza mutazione [ 27 , 31 ] non possa essere esclusa.
Cistinuria e consulenza genetica
Contrariamente ad altri disturbi caratterizzati da urolitiasi, la cistinuria è causata esclusivamente da mutazioni genomiche. Tuttavia, l’interpretazione dei risultati molecolari e la consulenza genetica nelle famiglie con cistinuria sono spesso complicate dalla difficoltà di differenziare pazienti e portatori eterozigoti di mutazioni con ambiguità cliniche. In queste situazioni, i pazienti e le loro famiglie chiedono spesso una previsione del decorso clinico o un regime terapeutico mirato. Tuttavia, una prognosi inequivocabile è semplicemente possibile a causa dell’ampia variabilità clinica anche nei portatori della stessa mutazione e l’influenza di fattori endo- ed esogeni finora sconosciuti che modificano. Tuttavia, una consulenza più diretta è diventata possibile sulla base della classificazione molecolare suggerita da DelloStrologo et al. [11 ]:
AA (omozigosi per una o eterozigosi composta per due mutazioni SLC3A1 ) è principalmente coerente con un’eredità autosomica recessiva della cistinuria. I genitori sono portatori obbligati ma con una normale escrezione renale di cistina, un rischio di ricorrenza del 25% può essere delineato per i bambini affetti da cistinuria. Tuttavia, ci sono prove che almeno la mutazione dupE5E9 è associata a cistinuria non di tipo I, vale a dire che i portatori mostrano un aumento dell’escrezione di cistina [ 10 ]. I portatori di eterozigoti di questa variante devono essere testati per l’escrezione urinaria di cistina.
UN? (eterozigosi per una mutazione SLC3A1 , non è stato possibile identificare una seconda mutazione): probabilmente è presente una seconda mutazione in uno dei due geni ma non rilevabile con i metodi applicati. Tuttavia, nel caso della diagnosi clinica di cistinuria, l’identificazione di una sola mutazione in SLC3A1 dovrebbe essere sufficiente per la conferma. Per stimare ulteriormente il rischio di ricorrenza in queste famiglie, è necessario determinare i modelli di escrezione urinaria e il genotipo della cistinuria dei genitori. Nel caso di una normale escrezione cistina in quel genitore che non contribuisce alla SLC3A1 eterozigosi composta mutazione per la identificato e uno sconosciuto SLC3A1la mutazione può essere delineata per il paziente. Se il genitore che non porta la mutazione SLC3A1 rilevata mostra un aumento dell’escrezione di cistina, si può ipotizzare una mutazione SLC7A9 . In tal caso, la previsione clinica della prole che si prevede sia eterozigote per le mutazioni (sconosciute) SLC7A9 è difficile a causa dell’ampia gamma di penetranza biochimica delle mutazioni in questo gene.
BB (omozigosi per una o eterozigosi composta per due mutazioni SLC7A9 ) è coerente con cistinuria. Il rischio di ricorrenza per fratelli e sorelle è almeno del 25%, ma a causa della possibile natura dominante della mutazione SLC7A9 e delle ampie gamme di escrezione di aminoacidi urinari anche nella stessa famiglia il rischio di sviluppare calcoli di cistina è maggiore. Qui l’analisi biochimica delle urine dei portatori della mutazione eterozigote e la conoscenza della natura patogena della mutazione potrebbero aiutare a delineare ulteriormente il rischio di sviluppare calcoli.
B? (eterozigosi per una mutazione SLC7A9 , non è stato possibile identificare una seconda mutazione): questa scoperta è la più difficile da interpretare: come discusso per il genotipo BB, la stessa mutazione potrebbe comportarsi in modo recessivo in una generazione e dominante in un’altra nella stessa famiglia . Quindi l’identificazione di una sola mutazione SLC7A9 potrebbe essere compatibile con un’escrezione di cistina estremamente aumentata, la prognosi nei neonati o nei bambini è semplicemente possibile.
AB (eterozigosi mista di una mutazione SLC3A1 e SLC7A9 ): questa scoperta spiega il fenotipo clinico, ma come discusso per A ?, BB e B? genotipi, il probabile effetto dominante di una mutazione SLC7A9 deve essere tenuto presente nella consulenza genetica.
AAA / AAB / ABB (tre mutazioni SLC3A1 / SLC7A9 in un paziente): questa rara scoperta spiega il fenotipo, ma i genitori dovrebbero essere controllati per le mutazioni per identificare il cromosoma che ospita due mutazioni e quindi per evitare risultati di test sui portatori falsi negativi in l’ulteriore famiglia.
Nella pratica clinica, i risultati dei test genetici molecolari influenzano appena la prognosi e la terapia della cistinuria poiché attualmente non esistono terapie causali. Infatti, oltre il 95% di tutti i portatori di due mutazioni SLC3A1 o SLC7A9 (genotipi AA, BB, AB) svilupperanno calcoli renali nella loro vita, ma l’età della formazione di calcoli renali è difficile da prevedere e mostra un’ampia variabilità intrafamiliale. Inoltre, il modello di escrezione urinaria nei portatori della mutazione eterozigote SLC7A9 è estremamente variabile e pertanto difficilmente consente una previsione del decorso clinico mentre la maggior parte degli eterozigoti SLC3A1 non presenta un fenotipo biochimico (per la revisione: [ 12]). Per quanto la conoscenza completa della fisiopatologia della cistinuria non possa essere applicata in una terapia causativa, la determinazione biochimica del modello di escrezione della cistina urinaria rimane lo strumento di base per la prognosi e la gestione terapeutica della cistinuria.
Tratto da:-
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-7-19