
La malattia
Che cos’è?
La retinite pigmentosa (RP) è una malattia genetica degenerativa dell’occhio caratterizzata da una perdita graduale e graduale della visione che di solito progredisce fino alla cecità. RP è anche chiamata distrofia cono- rodina o retinite pigmentosa , sinonimi derivati dal suo nome in inglese.La prevalenza (numero di casi in una data popolazione in un dato momento) è 1 su 3.500 persone.La malattia colpisce entrambi i sessi indipendentemente dalla loro origine geografica. Può iniziare a qualsiasi età con una frequenza di insorgenza maggiore tra 10 e 30 anni.
RP è una malattia genetica causata dal cambiamento (mutazione) di geni coinvolti nel funzionamento e regolazione delle cellule della retina, fotorecettori, imprescindibile per una visione.
Nella metà dei casi, la persona colpita è la prima a essere nella famiglia. Si dice che il suo caso sia sporadico. In questo caso la malattia è il risultato di un “incidente genetico”, ma questo cambiamento (mutazione) di un gene, che si verificano in modo imprevisto, è trasmissibile alla prole. Nell’altra metà dei casi, il PR è chiamato “famiglia” perché sono interessate almeno due persone della stessa famiglia. In tutti i casi, la trasmissione avviene in diversi modi: autosomica dominante, autosomica recessiva, trasmissione collegato alla X (vedi capitolo “Aspetti genetici”).
I geni responsabili sono molto numerosi, più di 39 fino ad oggi. L’alterazione di alcuni geni è più frequente. Nelle forme autosomica dominante, mutazione della rodopsina ( RHO ) si trova in uno su quattro, mentre una mutazione nel gene ri tinitis pigmentosa 1 ( RP1 è presente nel 6-8% dei casi). Retinitis pigmentosa GTPase regulator ( RPGR ) è il principale gene per le forme la cui trasmissione è legata al cromosoma X (vedere il capitolo “Aspetti genetici”).
RP di solito inizia con problemi di vista quando l’intensità della luce diminuisce (cecità notturna). Le difficoltà di adattamento all’oscurità sono frequenti, ad esempio durante il passaggio di una stanza molto illuminata verso una stanza buia.
Gradualmente, il campo visivo si restringe con l’impossibilità di vedere le cose su, giù o ai lati, dando un’impressione di “visione a tunnel”: ciò corrisponde a una riduzione della visione periferica. Questo attacco è bilaterale, entrambi gli occhi sono colpiti. La vita quotidiana viene gradualmente avvertita: presenza di un certo imbarazzo, difficoltà a guidare di notte, a volte anche giorno per mancanza di una visione globale della strada, frequenti oggetti a percussione durante la passeggiata …
I disturbi nella visione dei colori, in particolare blu e giallo, sono spesso presenti (discromatopsia). A volte una maggiore sensibilità ad alta intensità luminosa (fotofobia) può comparire in seguito.
La visione centrale è generalmente conservata fino alle fasi avanzate della malattia. La sua diminuzione si manifesta dapprima in difficoltà nell’esecuzione di attività meticolose o nella lettura, e progressivamente diminuisce l’acuità visiva e generalmente provoca cecità. In alcuni casi altri problemi oculari possono essere aggiunti all’RP, come l’edema maculare, e possono comportare una perdita della visione centrale prima del restringimento principale del campo visivo.
Altri problemi agli occhi si verificano più frequentemente nelle persone con RP rispetto alla popolazione generale come la miopia nelle forme di trasmissione legate all’X (scarsa visione a distanza) o l’astigmatismo significativo che si verifica spesso nelle forme precoci e tardive. grave (visione distorta distorta). La cataratta, che è una opacità della lente che distrugge progressivamente la visione, è anche una complicanza comune di RP.
Per le forme ereditarie, sembra esserci una relazione tra la modalità di trasmissione di RP e la gravità della malattia. Le forme di trasmissione autosomica dominante sono generalmente le forme meno gravi, l’evoluzione della malattia è più lenta, la visione centrale è preservata più a lungo. Inoltre, in queste forme, le manifestazioni di RP variano da un individuo all’altro (penetranza variabile). Le forme di ereditarietà autosomica recessiva iniziano di solito prima dei 20 anni e le forme legate all’X sono le forme più gravi, con un inizio più precoce.
C’è una malattia chiamata cono-astadistrofia che a volte viene compresa con il termine di RP. Questa malattia non presenta le stesse manifestazioni dell’RP: la visione centrale viene prima persa, poi la visione periferica.
Il PR può essere associato ad altre manifestazioni ed è quindi parte di diverse e diverse sindromi (tutte le manifestazioni fisiche). Il termine RP è a volte usato al plurale o associato per estensione a quello della retinopatia pigmentaria per designare questo gruppo eterogeneo di malattie genetiche che hanno in comune la presenza di un RP. Nella più frequente, la sindrome di Usher si trova in cui l’RP è associato alla sordità presente dalla nascita. Oltre alla RP, la sindrome di Bardet Biedl è associata a obesità, disabilità intellettiva, presenza di dita soprannumerarie (polidattilia) e malformazioni genitali e renali. Queste sindromi hanno in comune la presenza di PR ma sono diverse da PR “isolate” o talvolta non sindromiche.
La retina è un tessuto situato nella parte inferiore dell’occhio in cui si formano le immagini. Esso è diviso in due parti: il nervo retinico costituito da diversi milioni di cellule, le torécepteurs grafie fissano la luce, e uno strato sottile, l’epitelio pigmentato. I fotorecettori sono divisi in due tipi di cellule, coni e bastoncelli ( cono e astain inglese, rispettivamente). I coni lavorano alla luce del giorno e permettono al nostro occhio di distinguere i dettagli e percepire i colori. La parte centrale della retina o fovea è specializzata nella percezione dei dettagli ed è composta quasi esclusivamente da coni, mentre il resto della retina, compresa la macula, è in gran parte dominato da aste. Questi intervengono quando la luce diminuisce o in condizioni di scarsa illuminazione. La visione periferica è mediata dai coni in piena luce e dalle barre nella penombra.
In RP, è influenzata la capacità della retina di rispondere alla luce. Questa alterazione è dovuta alla graduale distruzione di bastoncelli e coni, che si traduce nella graduale comparsa di piccoli depositi pigmentati nella retina dando così il nome alla malattia e alle aree non visibili nel campo visivo denominate scotomi. La fusione di scotomie periferiche spiega questa impressione di visione a tunnel.

Figura 1: diagramma dell’occhio.
Qual è l’ evoluzione?
L’evoluzione dell’RP è variabile da persona a persona, ma in genere è piuttosto lenta e si estende per diversi decenni. Nonostante una continua evoluzione, può dare l’impressione di alternare lunghe fasi di stagnazione, seguite da fasi di rapido degrado. A volte le persone con RP possono mantenere una piccola parte del campo visivo fino alla vecchiaia, e alcune forme lievi o tardive non portano necessariamente alla completa cecità.
La diagnosi
Qual è la diagnosi?
Il più delle volte, la diagnosi viene fatta durante una valutazione oftalmologica eseguita a causa di problemi di visione notturna, un incidente dovuto alla mancanza di visione periferica o una diminuzione dell’acuità visiva. Ulteriori test confermeranno la diagnosi. L’elettroretinogramma (ERG) consente di esplorare l’attività della retina durante una stimolazione luminosa e mette in evidenza la disfunzione di bastoncelli e coni. La realizzazione di un ERG non richiede il ricovero in ospedale, è indolore ma a volte viene eseguita in anestesia locale. I meriti degli occhi con una piccola lampada (oftalmoscopio), dopo aver pupilla dilatata con collirio aproprié, metterà in evidenza la presenza di depositi pigmentate (scotoma) nella parte interessata. Un esame del campo visivo, centrale e periferico,
Altri test complementari possono essere eseguiti ma non sono obbligatori per la diagnosi di RP: l’elettrooculogramma (EOG) esplora l’attività delle cellule della retina. Il test di adattamento misura la capacità dell’occhio di adattarsi all’oscurità. I potenziali evocati visivi (PEV) esplorano l’attività del nervo ottico e dei percorsi visivi (fibre nervose che trasmettono informazioni dalla retina al cervello). Infine, una fluorangiografia può essere prescritto ed usato per scattare foto della parte posteriore dell’occhio: un colorante, la fluoresceina, viene iniettato nella vena del braccio e pochi secondi più tardi, arriva alla parte posteriore dell’occhio che è quindi osservato.
Quando viene effettuata la diagnosi di RP, il medico studierà se sono presenti altre anormalità per escludere qualsiasi altra malattia per cui la RP è una delle manifestazioni cliniche, come ad esempio la sindrome di Usher.
Come si differenzia?
In alcuni casi, la RP può verificarsi alla nascita o pochi mesi dopo e può essere confusa con l’amaurosi congenita di Leber. Inoltre, in entrambe le malattie sono presenti anomalie genetiche comuni. Può anche essere confuso con anomalie stazionarie del funzionamento della retina o anche con distrofie di coni (o distrofie di coni).), malattie caratterizzate solo da danni ai coni della retina. Il PR può essere confuso con la coroidemia, che si manifesta anche con una progressiva riduzione del campo visivo e delle macchie pigmentate nel fondo. Può essere confuso con la cosiddetta cecità notturna congenita stazionaria in cui la perdita della vista è limitata alla visione notturna. Infine, alcune infezioni o infiammazioni dell’occhio e l’avvelenamento da farmaci possono dare le stesse manifestazioni oculari dell’RP.
Quali sono i rischi delle persone a rischio prima che vengano segnalati?
Il test genetico è fattibile in familiari di una persona con RP familiare, ma solo quando l’anomalia genetica (mutazione) è stata identificata nella persona interessata. Consiste nel cercare i parenti a rischio (genitori, figli, fratelli) dell’anomalia genetica da a sangue, anche prima che compaiano i sintomi. Quindi, è uno studio di biologia molecolare che può rispondere a questa domanda cercando direttamente la mutazione nel gene coinvolto. Tuttavia, questa diagnosi predittiva o pre-sintomatica deve essere fatta solo rispettando un certo numero di principi, a causa delle sue implicazioni psicologiche. Questi test possono essere eseguiti solo nel contesto di consultazioni specializzate e multidisciplinari che coinvolgono genetisti e psicologi. I ritardi tra la prima consultazione e il campionamento per le analisi genetiche devono consentire al richiedente di poter eventualmente rinunciare a conoscere il suo stato. Questo ritardo, in alcuni casi, può raggiungere diversi mesi. Nessun test diagnostico viene eseguito alla prima visita. Il candidato al test deve essere maggiore e indipendente; deve firmare un modulo di consenso informato, vale a dire dare il suo consenso dopo aver ricevuto tutte le informazioni necessarie sul corso del test, le sue conseguenze e le possibili alternative. I risultati sono riservati e interessano solo le persone interessate. Tutte queste precauzioni sono prese in modo che le persone a rischio abbiano il tempo di pensare. Un seguito psicologico viene proposto dopo il rendering del test e questo è qualunque sia il risultato dal momento che a volte sono possibili conseguenze indesiderabili anche in caso di risultato favorevole. A volte è desiderabile effettuare una diagnosi precoce della malattia per consentire una gestione e un monitoraggio adeguati. deve firmare un modulo di consenso informato, vale a dire dare il suo consenso dopo aver ricevuto tutte le informazioni necessarie sul corso del test, le sue conseguenze e le possibili alternative. I risultati sono riservati e interessano solo le persone interessate. Tutte queste precauzioni sono prese in modo che le persone a rischio abbiano il tempo di pensare. Un seguito psicologico viene proposto dopo il rendering del test e questo è qualunque sia il risultato dal momento che a volte sono possibili conseguenze indesiderabili anche in caso di risultato favorevole. A volte è desiderabile effettuare una diagnosi precoce della malattia per consentire una gestione e un monitoraggio adeguati. deve firmare un modulo di consenso informato, vale a dire dare il suo consenso dopo aver ricevuto tutte le informazioni necessarie sul corso del test, le sue conseguenze e le possibili alternative. I risultati sono riservati e interessano solo le persone interessate. Tutte queste precauzioni sono prese in modo che le persone a rischio abbiano il tempo di pensare. Un seguito psicologico viene proposto dopo il rendering del test e questo è qualunque sia il risultato dal momento che a volte sono possibili conseguenze indesiderabili anche in caso di risultato favorevole. A volte è desiderabile effettuare una diagnosi precoce della malattia per consentire una gestione e un monitoraggio adeguati. I risultati sono riservati e interessano solo le persone interessate. Tutte queste precauzioni sono prese in modo che le persone a rischio abbiano il tempo di pensare. Un seguito psicologico viene proposto dopo il rendering del test e questo è qualunque sia il risultato dal momento che a volte sono possibili conseguenze indesiderabili anche in caso di risultato favorevole. A volte è desiderabile effettuare una diagnosi precoce della malattia per consentire una gestione e un monitoraggio adeguati. I risultati sono riservati e interessano solo le persone interessate. Tutte queste precauzioni sono prese in modo che le persone a rischio abbiano il tempo di pensare. Un seguito psicologico viene proposto dopo il rendering del test e questo è qualunque sia il risultato dal momento che a volte sono possibili conseguenze indesiderabili anche in caso di risultato favorevole. A volte è desiderabile effettuare una diagnosi precoce della malattia per consentire una gestione e un monitoraggio adeguati.
Se l’anomalia genetica non viene identificata, è possibile eseguire un elettroretinogramma (ERG) per rilevare la presenza di RP in altri membri della famiglia di una persona con RP. In effetti, può rivelare anormalità nei coni (bastoncelli) funzionanti (funzionali) prima della comparsa dei sintomi.
Aspetti genetici
Quali sono i rischi di comunicare con i bambini?
La PR viene trasmessa in modi molto diversi (eterogeneità genetica), il rischio di trasmissione ai bambini varia a seconda del tipo di ereditarietà. Circa il 15-20% di RP familiare è autosomico dominante, il 20-25% è autosomico recessivo e il 10-15% è legato all’X. Infine, alcune mutazioni del DNA mitocondriale si trovano anche in alcuni casi di RP.
Trasmissione autosomica dominante
( figura 2 )
Quando la malattia è definita autosomica dominante, significa che può essere trasmessa di generazione in generazione. Una persona a rischio ha il rischio di trasmettere la malattia ai propri figli in ogni gravidanza, indipendentemente dal sesso: una singola copia del gene mutato, trasmessa dalla madre o dal padre, causa l’insorgenza della malattia.
Trasmissione di tipo autosomico recessivo
( Figura 3 )
Quando la malattia è definita autosomica recessiva, sono interessati solo i bambini che hanno ricevuto il gene alterato (gene mutato) sia dal padre che dalla madre. Pertanto, i pazienti infetti trasportano due copie del gene mutato (si dice che siano omozigoti, quando i geni mutati sono compositi identici o eterozigoti, quando i due geni mutati sono diversi) mentre ciascuno dei genitori è solo un portatore solo una copia (sono chiamati eterozigoti). In questo caso i genitori sono portatori sani, non mostrano i segni della malattia. Questa malattia di solito colpisce solo i fratelli della stessa famiglia. La probabilità di avere un altro figlio è una su quattro.

Figura 2
Trasmissione autosomica dominante
Uno dei genitori ha una copia mutata del gene (A) e ha la malattia, proprio come suo figlio A / a. la
in ogni gravidanza, il rischio che un bambino di una persona A / a sia malato è del 50%.
I bambini a / a non sono malati e non possono trasmettere la malattia (portano due copie normali del gene a / a).

Figura 3
Ereditarietà autosomica recessiva
Entrambi i genitori portano una copia del gene mutato (a) e una copia del gene normale (A): non sono malati
(hanno detto che sono eterozigoti).
Il bambino ha recuperato i due geni mutati di suo padre e sua madre: ha la retinite pigmentosa (si dice che sia omozigote).
I bambini A / a portano il gene, sono eterozigoti: non svilupperanno la malattia, ma possono trasmettere il gene come genitori. Sono chiamati portatori sani.
Il bambino A / A non ha recuperato il gene mutato da sua madre o suo padre: non è malato e non è a rischio di trasmettere la malattia.
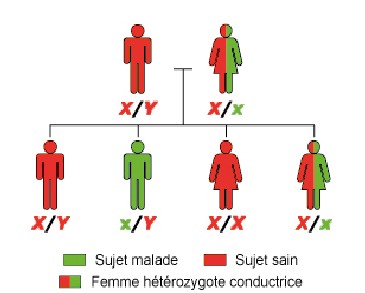
Eredità recessiva legata all’X ( Figura 4 )
Quando si dice che la malattia è recessiva legata all’X, significa che colpisce principalmente i maschi. La donna che trasporta un gene alterato su uno dei suoi cromosomi X di solito non ha manifestazioni della malattia; il gene inalterato sul secondo cromosoma X compensa questo difetto. D’altra parte, un bambino maschio che ha lo stesso difetto sull’unico cromosoma X che possiede (e ereditato da sua madre) svilupperà la malattia. Una donna eterozigote (o conduttrice) può dare alla luce un ragazzo malato una seconda volta; se nessuna delle sue figlie è malata, ma una su due può essere d’altra parte conduttrice come sua madre e può avere un figlio colpito.
Un uomo malato avrà solo bambini incolumi. Tuttavia, tutte le sue figlie sono costruttivamente conduttive dal momento che ricevono dal loro padre il loro cromosoma X che trasporta il gene difettoso. D’altra parte, tutti i suoi figli che ereditano il cromosoma Y, non coinvolti nella malattia, sono illesi.
Se il padre ha PR e la madre è un autista, allora tutte le ragazze avranno un cromosoma X mutato, quello del padre. Coloro che hanno ricevuto due copie del cromosoma mutato saranno malati. Tutti i ragazzi avranno un mezzo rischio di essere colpiti ricevendo il cromosoma X mutato dalla madre eterozigote.
In caso di una nuova mutazione nel bambino affetto (neomutazione), alcune madri di bambini malati non sono conduttive; il rischio riguarderà in seguito i figli non ancora nati delle figlie di quest’ultimo.
Figura 4
Trasmissione X-linked
Ad ogni gravidanza, il rischio:
– un ragazzo è malato è del 50%
– che una ragazza è malata è del 50%. Le ragazze sono meno gravemente colpite rispetto ai ragazzi.
Altre modalità di trasmissione molto più rare
– Molto più raramente, l’anomalia genetica può essere di origine mitocondriale. In questo caso, le donne malate trasmettono la malattia a tutti i loro figli indipendentemente dal sesso, ma gli uomini malati non trasmettono la malattia a nessuno dei loro figli.
Infine, anche in casi molto rari, la RP è associato uniparental disomy: il paziente ha due cromosomi dallo stesso genitore, invece di avere una materna e una paterna origine. Nel RP, il bambino riceve due cromosomi identici (copia l’uno dall’altro e che trasportano il gene mutato), uno dei suoi genitori (isodisomie uniparentale). Questa modalità di trasmissione rimane eccezionale.
In tutti i casi, è altamente consigliabile per fissare un appuntamento per la consulenza genetica (in un centro medico della genetica) per la valutazione del rischio accurata e di ricevere spiegazioni appropriate soprattutto il desiderio di gravidanza.
Qual è la diagnosi?
La diagnosi prenatale consiste nel cercare l’anormalità genetica sulle cellule prodotte dal feto. Le due tecniche utilizzate sono l’amniocentesi e il campionamento dei villi coriali. L’amniocentesi viene utilizzata per esaminare le cellule galleggianti nel fluido che circonda il feto (liquido amniotico) per cercare l’anormalità genetica che causa la malattia. Il campione viene prelevato in anestesia locale usando una siringa. Questo esame è offerto al 15 ° settimana di gravidanza.
La rimozione dei villi coriali ha il vantaggio di essere eseguita prima in gravidanza: consiste nel prelevare una quantità molto piccola di tessuto all’origine della placenta (il trofoblasto) all’esterno della busta dove il feto si sviluppa Il test viene generalmente fatto mento al 11 ° settimana di gravidanza.
Questi esami comportano un basso rischio di aborto, diverso a seconda della scelta della tecnica di campionamento, che dovrebbe essere discusso previa consultazione della genetica.
Mentre una specifica mutazione in RP è evidenziata dalla diagnosi prenatale, è ancora molto difficile predire l’età di esordio e la velocità di progressione della RP. In effetti, le manifestazioni della malattia sono molto variabili, anche tra le persone che portano la stessa mutazione genetica.
Quali sono i rischi per la tua famiglia?
Se un membro della famiglia è interessato, vi è il rischio che altri siano interessati, anche se si tratta di un caso isolato. In quest’ultimo caso, la mutazione è successivamente trasmissibile alla prole. Questo è il motivo per cui è importante informare gli altri membri della famiglia una volta fatta la diagnosi, in modo che possano, se lo desiderano, essere esaminati.
Trattamento, cura, prevenzione
Esiste un trattamento per questa malattia?
Non esiste attualmente una cura per RA.
Alcune precauzioni possono rallentare la progressione della malattia. Si raccomanda l’uso di lenti protettive e filtranti adeguate che proteggano dalla luce e dalla luce ultravioletta. Il loro obiettivo è principalmente quello di ridurre la sensazione di abbagliamento, proprio come indossare un cappello visiera. Si consiglia inoltre di evitare l’esposizione al sole senza questa protezione (mare, montagna).L’assunzione di vitamina A ed E può rallentare il danno delle cellule coinvolte, coni e bastoncini. Questo effetto benefico è ancora molto discusso dalla comunità medica e scientifica. Se viene proposto questo trattamento, un semplice dosaggio del sangue di trigliceridi, enzimi epatici e retinolo plasmatico viene eseguito mediante semplice esame del sangue. Inoltre, la vitamina A è una fonte di malformazione nel feto, la sua prescrizione nelle donne in età fertile deve essere più particolarmente monitorata.In caso di cataratta, si consiglia la chirurgia. Questa operazione non è diversa da quella eseguita in persone che non hanno RP.
Quali sono le altre condizioni di salute per questa malattia?
Nonostante l’assenza di trattamenti curativi, quando si raggiunge la visione centrale è possibile offrire diversi ausili per “ipovedenti”. In effetti, la visione può essere spesso migliorata con dispositivi speciali. Si tratta di ausili ottici come lenti d’ingrandimento, lenti d’ingrandimento, telescopi o ausili non ottici costituiti da una serie di articoli che possono facilitare le attività della vita quotidiana: libri e riviste con caratteri grandi. res, giocando a carte con numeri grandi, quadranti telefonici e calcolatrici di grande formato, ad esempio orologi parlanti. Infine, gli aiuti elettronici come i sistemi televisivi a circuito chiuso con dispositivi di ingrandimento e dispositivi di lettura computerizzati integrati sono utili in alcune circostanze.
Cosa fare?
Il follow-up è fatto da un oftalmologo. Si raccomanda inoltre di incontrare un genetista che conosce le malattie degli occhi. Questo medico sarà in grado di spiegare ai genitori la modalità di trasmissione della malattia e i rischi a cui sono esposti i membri della famiglia e le opzioni a loro disposizione.
Quali sono le conseguenze dell’istruzione familiare, professionale, sociale, scolastica, sportiva e familiare?
Nell’RP, la perdita della vista è progressiva, la persona deve quindi adattare le sue abitudini quotidiane in base alle sue capacità visive. Le abilità visive compromesse possono avere un impatto significativo sulla vita dei bambini con RP, in particolare la loro istruzione scolastica, che a volte non può continuare nelle normali situazioni scolastiche. L’orientamento professionale dovrebbe anche essere adattato alle capacità visive. RP è generalmente incompatibile con la guida.
L’assistenza psicologica è molto utile per ottimizzare l’integrazione socioprofessionale e per imparare a convivere con una malattia progressiva che può portare alla cecità.
Dov’è la ricerca?
Attualmente, i ricercatori mirano a individuare tutti i geni responsabili di PR e a comprendere meglio i meccanismi coinvolti nelle manifestazioni della malattia per migliorarne il trattamento. Diversi approcci terapeutici sono anche oggetto di numerosi lavori: terapia genica, fattori neutrofici, cellule staminali, retina artificiale, ripristino delle funzioni visive mediante trapianto di cellule della retina e trattamenti farmacologici. Tuttavia, ad oggi, questi approcci sono ancora lontani dalle applicazioni terapeutiche per il trattamento della RP.
Tratto da:- www.orpha.net/data/patho/Pub/fr/RetinitePigmentaire-FRfrPub659v01.pdf | Mai 2007